ChatGPT在生物信息学领域的应用与局限
文章主题:生物信息学, ChatGPT, 对话形式, 表达量分析
近期,我们的生物信息学交流群中,ChatGPT成为了热门话题。无论是简要的试用体验分享,还是深入的焦虑分析以及行业和国家大事的探讨,都使得这个群体充满了活力。这种现象很自然地让我们联想到当前的流量竞争环境,人们往往会追求更高的阅读量,因此我相信这篇文章也将取得不错的关注度。
我看了看朋友们秀出来的跟ChatGPT的生物信息学相关对话,起初还以为是很高大上。比如:
构建一个R语言里面的S4对象(高级数据结构)
ChatGPT 的强大之处在于其能够理解我们提出的问题,这一点确实令人印象深刻。在它的回答中,它首先详细解释了 S4 对象(高级数据结构),并且随后给出了相应的示例,这无疑为我们提供了极大的帮助。

然而,一旦你熟悉了如何查阅该对象的帮助文档——即help函数,你就能非常清晰地了解到关于此对象的全面介绍,并且其中还包含有示例代码的展示。
# create an instance of ExpressionSetExpressionSet()
ExpressionSet(assayData=matrix(runif(1000), nrow=100, ncol=10))
# update an existing ExpressionSetdata(sample.ExpressionSet)
updateObject(sample.ExpressionSet)
# information about assay and sample datafeatureNames(sample.ExpressionSet)[1:10]
sampleNames(sample.ExpressionSet)[1:5]
experimentData(sample.ExpressionSet)
# subset: first 10 genes, samples 2, 4, and 10expressionSet <- sample.ExpressionSet[1:10,c(2,4,10)]
# named features and their expression levelssubset <- expressionSet[c(“AFFX-BioC-3_at”,“AFFX-BioDn-5_at”),]
exprs(subset)
# samples with above-average score in phenoData在数据处理过程中,我们经常需要对一些数值指标进行分析和评估。其中, highScores 和 mean(expressionSet$score) 是两个常用的数值。通过比较 highScores 与 mean(expressionSet$score) 的关系,我们可以更好地了解数据集中的表现情况。
expressionSet[,highScores]
# (automatically) coerce to data.frame在撰写本文时,我们采用了lm()函数来分析数据集 subset,该函数的因变量是score,自变量是AFFX.BioDn.5_at和AFFX.BioC.3_at。通过这种方式,我们可以更好地理解这些变量之间的关系,并从中得出有价值的结论。
ChatGPT 的技术创新在于将官方文档以对话形式呈现给读者,这确实需要一定的技术实力。不过,我目前还不确定它是从其他来源直接复制粘贴过来,还是经过了一定的处理和转换。无论如何,这种新颖的呈现方式无疑能够增加文档的可读性和吸引力。
差异分析
基于表达量矩阵的差异分析过程可以看我8年前的芯片教程,推文在:
解读GEO数据存放规律及下载,一文就够解读SRA数据库规律一文就够从GEO数据库下载得到表达矩阵 一文就够GSEA分析一文就够(单机版+R语言版)根据分组信息做差异分析- 这个一文不够的差异分析得到的结果注释一文就够针对不同表达量矩阵格式,里面有大量的包可以选择,如果是表达量芯片我们默认的是limma,而如果是转录组测序的counts矩阵格式,我们会使用DESeq2,edgeR等等,那么让我们看看ChatGPT的回答:
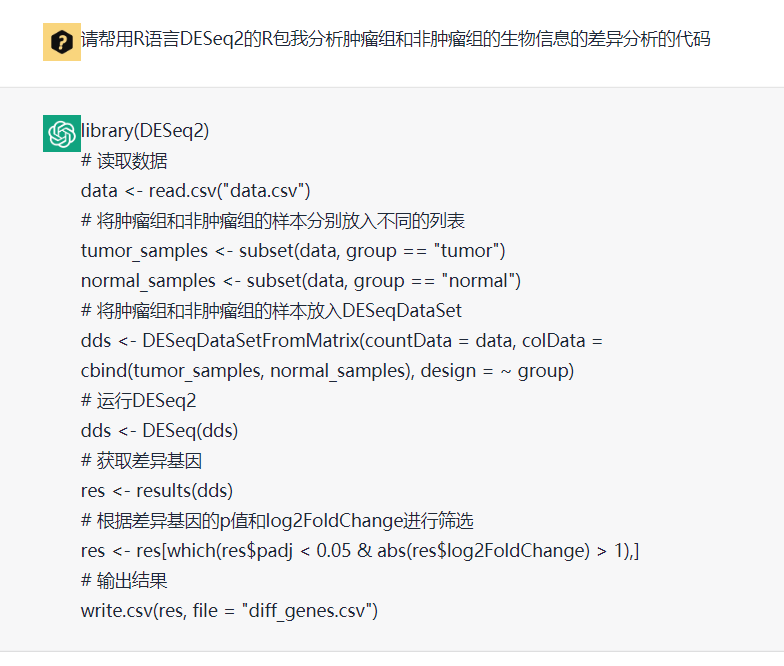
实际上,就是把这个包的文档给你了,很简单的看文档:
library(DESeq2)
vignette(DESeq2)
当然了,包的官方文档确实是又臭又长,所以ChatGPT做了一个精简,让初学者可以一目了然使用起来,但是忽略了大量的背后的参数和原理,以及其它案例,大概率上还是需要去读文档。。。
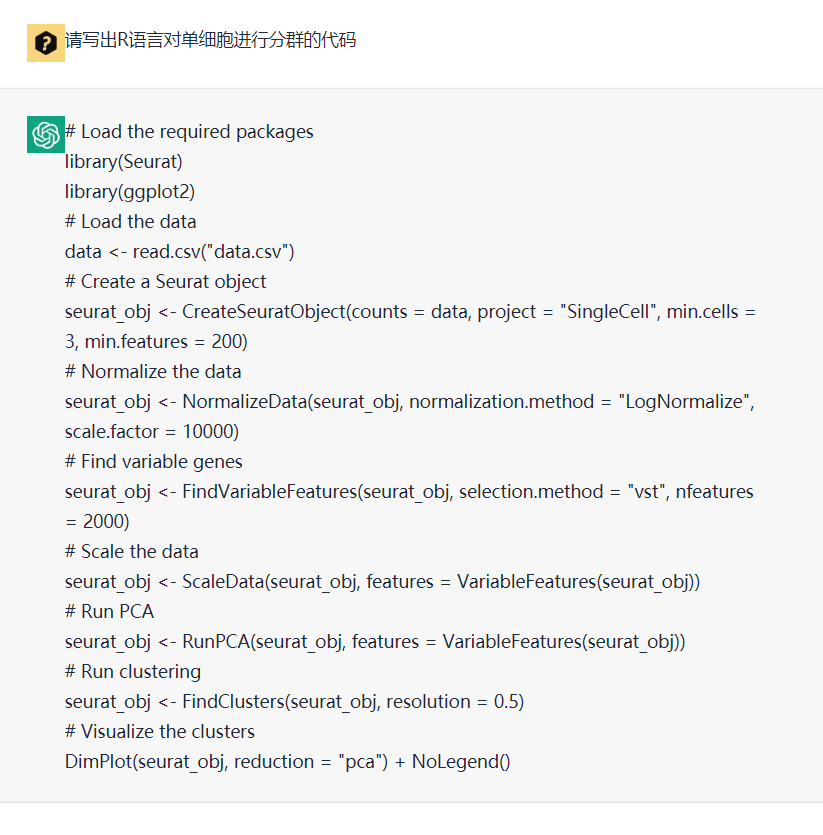
单细胞基础流程
就是拿到了单细胞转录组表达量矩阵文件之后,走单细胞转录组流程即可, 可以做harmony或者CCA的整合,然后降维聚类分群,如果你对单细胞数据分析还没有基础认知,可以看基础10讲:
01. 上游分析流程02.课题多少个样品,测序数据量如何03. 过滤不合格细胞和基因(数据质控很重要)04. 过滤线粒体核糖体基因05. 去除细胞效应和基因效应06.单细胞转录组数据的降维聚类分群07.单细胞转录组数据处理之细胞亚群注释08.把拿到的亚群进行更细致的分群09.单细胞转录组数据处理之细胞亚群比例比较事实上无论是Seurat自己的官方文档,还是我们公众号的介绍,相关基础代码都是足够丰富了,而ChatGPT无非就是代替你查文档罢了。

其实还不如直接搜索指定公众号(生信技能树)推文
我一直强调,【先搜索后提问】,我把大概1.3万篇笔记都分享在公众号里面了!告诉你如何去搜索我们生信技能树公众号教程,自行点学会在技能树公众号历史教程里面根据关键词查询,基本上初学者问题都有解决方案!你学会搜索,然后尝试着先搜索你的问题。并且强调大家【搜索完毕告诉我你的关键词,以及微信发给我你搜索的最佳结果推文】
如果是在你的手机里面的微信,下面的三个步骤即可:
要搜索自己关注的某个公众号(生信技能树)的文章,需要先点击进入该公众号,然后点击右上角的三个点按钮。(设置为星标)其实右上角的三个点按钮按钮旁边就是一个迷你版本的放大镜,就是可以点击的搜索框,输入需要搜索的关键词后点击输入法上的【搜索】按钮,就会出现带有该关键词的结果,这些文章都是这个公众号曾经发布的内容。如果是使用电脑,那么打开浏览器到搜狗微信搜索:https://weixin.sogou.com/
然后输入不同关键词即可:
差异分析 “生信技能树”甲基化 “生信技能树”单细胞 “生信技能树”文末友情宣传强烈建议你推荐给身边的博士后以及年轻生物学PI,多一点数据认知,让他们的科研上一个台阶:
生物信息学马拉松授课(买一得五) ,你的生物信息学入门课144线程640Gb内存服务器共享一年仍然是仅需800千呼万唤始出来的独享生物信息学云服务器
AI时代,拥有个人微信机器人AI助手!AI时代不落人后!
免费ChatGPT问答,办公、写作、生活好得力助手!
搜索微信号aigc666aigc999或上边扫码,即可拥有个人AI助手!